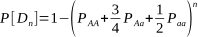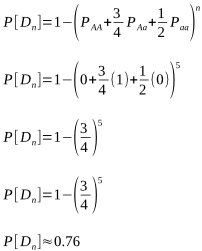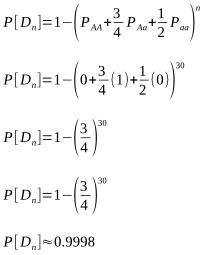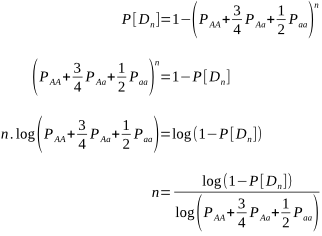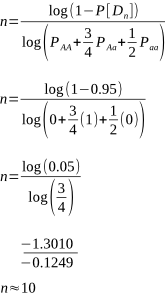To select for a simply-inherited trait requires knowing just three things: the number of loci involved (often just one), the number of alleles at each locus (usually a small number), and the genotypes or possible genotypes of the parents-to-be (again typically a small number).
In the case of a simply-inherited trait that is partially dominant, such as Andalusian chicken colour, all three pieces of information are known. There is just one locus (B), two alleles (’B’ and ‘b’), and three genotypes easily identifiable by eye (’BB’, black; ‘Bb’, slate blue; and ‘bb’, white).
Selection of a partially dominant trait is quite straightforward. Should you wish all white chickens, remove the blacks and blues from your flock and breed only the whites. All subsequent chicks will be white. Should you want only black chickens, remove the other two colours and breed only the blacks to produce generations of black-only birds. Keeping only slate-blue birds will produce all three genotypes in the next generation — not ideal if you don’t want black and white chickens, but at least you can still know their genotypes by looking at them. And to breed for a guaranteed all-blue next generation, simply cross black birds with white ones, and repeat.
Selecting for or against a genotype of a partially dominant trait is easy, simply because the genotype is visibly expressed.
What about a completely dominant trait, like the suri/huacaya genotype in alpacas mentioned last week?
Assume a herd of heterozygous suris, homozygous suris, and huacayas. The huacaya geneotype is recessive, so selecting for all huacayas and removing all suris will give you an all-huacaya herd that breeds true-to-type in no time. But should you want only suris, removing all huacayas won’t help, as the heterozygous suris are carriers of the huacaya allele, and may well produce future huacayas. They won’t always breed true-to-type.
It’s quite easy to select for or against a co-dominant genotype, as with the Andalusian chickens. It’s also quite easy to select for a recessive genotype, as with the huacaya alpacas. This is because you know without question the genotypes of the parents-to-be, and can predict the outcomes definitively.
It is more difficult to select against a recessive trait, ie to completely remove the recessive allele from a population. (Unless random genetic drift does it for you, but that is hardly a reliable breeding tool!) And likewise for the opposite side of the same coin: selecting for a homozygous dominant trait to again completely remove the recessive allele.
This is because the genotypes of the parents-to-be isn’t always known definitively. The recessive allele could be carried by any number of animals, though not expressed. Barring the existence of a genetic test, those carriers can’t be identified unless and until an offspring with the recessive phenotype is born.
Traditionally, a test mating was done only for males, partly because females aren’t usually as valuable to justify the test (just let the progeny ‘chips’ fall as they may, essentially), and partly because males can produce greater numbers of progeny, and more quickly, than a female. Apart from these practicalities, there is no intrinsic difference in testing males versus females. And the advent of embryo transfer (ET) technology does now allow greater numbers of offspring from a female to be tested, if the cost can be justified.
Revisiting the test cross scenarios of suri (dominant allele) over huacaya (recessive allele) last week, a suri need produce just one huacaya to show conclusively it is heterozygous. A homozygous suri never will:

© Optimate Group Pty Ltd
But after how many matings can we be sure an animal really is homozygous? It could be entirely due to chance that the recessive allele was never passed onto the progeny, despite numerous matings. Of course, the more matings that don’t produce the recessive phenotype, the higher the probability of homozygosity. If the probability is high enough we can conclude that an animal is indeed not a carrier.
Alpacas, as with other animals such as horses and cattle, are animals that typically have single births. Species where twinning or litters are the norm, such as with sheep, dogs, pigs and poultry, wouldn’t require as many matings for a homozygous recessive to appear.
The probabilities of different mating scenarios can be calculated, and we shall cover these in subsequent posts!



 distribution")